Jakie są standardowe sposoby obliczania odległości między poszczególnymi zapytaniami?
Zadałem podobne pytanie, pytając o odległość między „dokumentami” (artykuły z Wikipedii, wiadomości itp.). Zrobiłem to osobnym pytaniem, ponieważ zapytania wyszukiwania są znacznie mniejsze niż dokumenty i są znacznie głośniejsze. Dlatego nie wiem (i wątpię), czy zostaną tutaj użyte te same metryki odległości. Preferowane są albo waniliowe, leksykalne metryki odległości, albo najnowocześniejsze semantyczne metryki odległości, przy czym bardziej preferowane są te drugie.
Z mojego doświadczenia wynika, że tylko niektóre klasy zapytań można podzielić na cechy leksykalne (ze względu na niejednoznaczność języka naturalnego). Zamiast tego możesz spróbować użyć logicznych wyników wyszukiwania (witryn lub segmentów witryn, a nie dokumentów, bez rankingu) jako funkcji klasyfikacji (zamiast słów). To podejście sprawdza się dobrze w klasach, w których zapytanie zawiera dużą niejednoznaczność leksykalną, ale istnieje wiele dobrych witryn związanych z zapytaniem (np. Filmy, muzyka, zapytania reklamowe itp.).
Ponadto w przypadku klasyfikacji offline można wykonać LSI w macierzy witryny zapytań. Szczegółowe informacje można znaleźć w książce „Wprowadzenie do wyszukiwania informacji”.
Miara podobieństwa cosinusowego wykonuje dobrą (jeśli nie idealną) pracę przy kontrolowaniu długości dokumentu, więc porównanie podobieństwa 2 dokumentów lub 2 zapytań przy użyciu metryki cosinus i wag tf idf dla słów powinno działać dobrze w obu przypadkach. Poleciłbym również najpierw wykonać LSA na wagach tf idf, a następnie obliczyć cosinus odległość \ podobieństwa. Jeśli próbujesz zbudować wyszukiwarkę, polecam skorzystanie z bezpłatnej wyszukiwarki open source, takiej jak solr lub wyszukiwanie elastyczne, lub po prostu bibliotek surowego lucene, ponieważ wykonują większość pracy za Ciebie i mają dobre wbudowane metody obsługa zapytania w celu udokumentowania problemu podobieństwa.
Najlepsza biblioteka Pythona dla sieci neuronowych
Używam sieci neuronowych do rozwiązywania różnych problemów związanych z uczeniem maszynowym. Używam Pythona i pybrain, ale ta biblioteka jest prawie wycofana. Czy w Pythonie są inne dobre alternatywy?
AKTUALIZACJA: krajobraz nieco się zmienił, odkąd odpowiedziałem na to pytanie w lipcu 2014 roku, a w kosmos pojawiło się kilku nowych graczy. W szczególności polecam sprawdzić:
Lasagne: https://github.com/Lasagne/Lasagne
Keras: https://github.com/fchollet/keras
Deepy: https://github.com/uaca/deepy
Nolearn: https://github.com/dnouri/nolearn
Bloki: https://github.com/mila-udem/blocks
TensorFlow: https://github.com/tensorflow/tensorflow
Każdy z nich ma swoje mocne i słabe strony, więc daj im szansę i zobacz, który najlepiej pasuje do Twojego przypadku użycia. Chociaż rok temu poleciłbym używanie pylearn2, społeczność nie jest już aktywna, więc poleciłbym poszukać gdzie indziej. Moja pierwotna odpowiedź na odpowiedź znajduje się poniżej, ale w tym momencie jest ona w dużej mierze nieistotna. Pylearn2 jest ogólnie uważany za bibliotekę z wyboru dla sieci neuronowych i głębokiego uczenia się w Pythonie. Został zaprojektowany z myślą o łatwych eksperymentach naukowych, a nie łatwości użytkowania, więc krzywa uczenia się jest dość stroma, ale jeśli nie spiesz się i zastosujesz samouczki
Myślę, że będziesz zadowolony z jego funkcji. Zapewnione jest wszystko, od standardowych perceptronów wielowarstwowych, przez maszyny Boltzmanna z ograniczeniami, po sieci splotowe, po autenkodery. Zapewnia świetne wsparcie dla GPU i wszystko jest oparte na Theano, więc wydajność jest zazwyczaj całkiem dobra. Źródło Pylearn2 jest dostępne na github. Należy pamiętać, że Pylearn2 ma obecnie przeciwny problem do pybrain – zamiast zostać porzuconym, Pylearn2 jest w trakcie aktywnego rozwoju i podlega częstym zmianom.
Tensor Flow (docs) od Google to kolejny fajny framework, który ma automatyczne różnicowanie. Zapisałem kilka krótkich przemyśleń na temat Google Tensor Flow na moim blogu, wraz z przykładem MNIST, który mają w swoim samouczku. Lasagne (dokumentacja) jest bardzo fajna, ponieważ wykorzystuje theano (→ możesz użyć GPU) i sprawia, że jest prostszy w użyciu. O ile wiem, autor lasagne wygrał wyzwanie Galaktyki Kaggle. Miło jest, gdy się nie uczysz. Oto przykładowa sieć MNIST:
#!/usr/bin/env python
import lasagne
from lasagne import layers
from lasagne.updates import nesterov_momentum
from nolearn.lasagne import NeuralNet
import sys
import os
import gzip
import pickle
import numpy
PY2 = sys.version_info[0] == 2
if PY2:
from urllib import urlretrieve
def pickle_load(f, encoding):
return pickle.load(f)
else:
from urllib.request import urlretrieve
def pickle_load(f, encoding):
return pickle.load(f, encoding=encoding)
DATA_URL = ‘http://deeplearning.net/data/mnist/mnist.pkl.gz’
DATA_FILENAME = ‘mnist.pkl.gz’
def _load_data(url=DATA_URL, filename=DATA_FILENAME):
“””Load data from `url` and store the result in `filename`.”””
if not os.path.exists(filename):
print(“Downloading MNIST dataset”)
urlretrieve(url, filename)
with gzip.open(filename, ‘rb’) as f:
return pickle_load(f, encoding=’latin-1′)
def load_data():
“””Get data with labels, split into training, validation and test set.”””
data = _load_data()
X_train, y_train = data[0]
X_valid, y_valid = data[1]
X_test, y_test = data[2]
y_train = numpy.asarray(y_train, dtype=numpy.int32)
y_valid = numpy.asarray(y_valid, dtype=numpy.int32)
y_test = numpy.asarray(y_test, dtype=numpy.int32)
return dict(
X_train=X_train,
y_train=y_train,
X_valid=X_valid,
y_valid=y_valid,
X_test=X_test,
y_test=y_test,
num_examples_train=X_train.shape[0],
num_examples_valid=X_valid.shape[0],
num_examples_test=X_test.shape[0],
input_dim=X_train.shape[1],
output_dim=10,
)
def nn_example(data):
net1 = NeuralNet(
layers=[(‘input’, layers.InputLayer),
(‘hidden’, layers.DenseLayer),
(‘output’, layers.DenseLayer),
],
# layer parameters:
input_shape=(None, 28*28),
hidden_num_units=100, # number of units in ‘hidden’ layer
output_nonlinearity=lasagne.nonlinearities.softmax,
output_num_units=10, # 10 target values for the digits 0, 1, 2, …, 9
# optimization method:
update=nesterov_momentum,
update_learning_rate=0.01,
update_momentum=0.9,
max_epochs=10,
verbose=1,
)
# Train the network
net1.fit(data[‘X_train’], data[‘y_train’])
# Try the network on new data
print(“Feature vector (100-110): %s” % data[‘X_test’][0][100:110])
print(“Label: %s” % str(data[‘y_test’][0]))
print(“Predicted: %s” % str(net1.predict([data[‘X_test’][0]])))
def main():
data = load_data()
print(“Got %i testing datasets.” % len(data[‘X_train’]))
nn_example(data)
if __name__ == ‘__main__’:
main()
Caffe jest biblioteką C ++, ale ma powiązania z Pythonem. Większość rzeczy możesz zrobić za pomocą plików konfiguracyjnych (prototxt). Ma wiele opcji i może również korzystać z GPU.
Pylearn polega na Theano i jak wspomniano w drugiej odpowiedzi, korzystanie z biblioteki jest dość skomplikowane, dopóki go nie zdobędziesz. W międzyczasie sugerowałbym skorzystanie z Theanets. Jest również zbudowany na szczycie Theano, ale jest znacznie łatwiejszy w obsłudze. Może to prawda, że nie ma wszystkich funkcji Pylearn, ale do podstawowej pracy jest wystarczający. Jest to również oprogramowanie typu open source, więc jeśli się odważysz, możesz dodawać niestandardowe sieci w locie. EDYCJA: grudzień 2015 r. Ostatnio zacząłem używać Keras. Jest to nieco niższy poziom niż Theanets, ale znacznie mocniejszy. Do podstawowych testów odpowiedni jest Theanets. Ale jeśli chcesz przeprowadzić badania w dziedzinie ANN, Keras jest znacznie bardziej elastyczny. Dodatkowo Keras może używać Tensorflow jako zaplecza.
Wydajne dynamiczne grupowanie
Mam zestaw punktów danych z przedziału jednostkowego (tj. 1-wymiarowy zbiór danych z wartościami liczbowymi). Otrzymuję dodatkowe punkty danych online, a ponadto wartość niektórych punktów danych może zmieniać się dynamicznie. Szukam idealnego algorytmu grupowania, który
może skutecznie rozwiązać te problemy. Wiem, że sekwencyjne grupowanie k-średnich radzi sobie z dodawaniem nowych instancji i przypuszczam, że przy niewielkich modyfikacjach może działać z dynamicznymi wartościami instancji (tj. Najpierw pobiera zmodyfikowaną instancję z odpowiedniego klastra, następnie aktualizuje średnią klastra i na koniec podaje zmodyfikowana instancja jako dane wejściowe do algorytmu, tak jak dodanie niewidocznej instancji). Moim problemem związanym z użyciem algorytmu k-średnich jest wymóg podania liczby klastrów jako danych wejściowych. Wiem, że pokonują inne algorytmy grupowania (GA, MST, metody hierarchiczne itp.) Pod względem złożoności czasowo-przestrzennej. Szczerze mówiąc, nie jestem pewien, ale może uda mi się użyć jednego z wyżej wymienionych algorytmów. Nawet jeśli moje zbiory danych są stosunkowo duże, istnienie jednego wymiaru sprawia, że zastanawiam się. Dokładniej, typowy mój przypadek testowy zawierałby około 10 000-200 1-wymiarowych punktów danych. Chciałbym zakończyć grupowanie najlepiej w ciągu sekundy. Zakłada się, że dynamiczne zmiany punktów wartości są płynne, tj. Stosunkowo niewielkie. Dlatego wysoce preferowana jest możliwość korzystania z istniejących rozwiązań (tj. Możliwość kontynuowania klastrowania na istniejącym, gdy wartość jest zmieniana lub dodawana jest nowa). Podsumowując:
Czy możesz pomyśleć o algorytmie, który zapewni najlepszy punkt między wydajnością obliczeniową a dokładnością klastrów wrt. problem zdefiniowany powyżej?
Czy jest jakaś fajna heurystyka dla algorytmu k-średnich do automatycznego obliczania wartości K wcześniej?
Myślę, że hierarchiczne grupowanie byłoby bardziej wydajne w twoim przypadku (z jednym wymiarem). W zależności od zadania możesz zaimplementować coś takiego: Posiadanie N punktów danych di z ich 1-wymiarową wartością x <sup> i </sup>:
- Sortuj punkty danych na podstawie ich wartości x <sup> i </sup>.
- Oblicz odległości między sąsiednimi punktami danych (odległości N-1). Każdej odległości należy przypisać parę oryginalnych punktów danych (d <sup> i </sup>, d <sup> j </sup>).
- Posortuj odległości w porządku malejącym, aby wygenerować listę par punktów danych (d <sup> i </sup>, d <sup> j </sup>), zaczynając od najbliższej.
- Iteracyjnie łącz punkty danych (d <sup> i </sup>, d <sup> i </sup>) w klastry, zaczynając od początku listy (najbliższa para). (W zależności od aktualnego stanu di i dj, połączenie ich oznacza: (a) utworzenie nowego klastra dla dwóch nieklastrowanych punktów danych, (b) dodanie punktu danych do istniejącego klastra oraz (c) połączenie dwóch klastrów.)
- Przestań się łączyć, jeśli odległość przekracza pewien próg.
- Utwórz pojedyncze klastry dla punktów danych, które nie dostały się do klastrów.
Ten algorytm implementuje grupowanie pojedynczych powiązań. Można go łatwo dostroić, aby wdrożyć przeciętne połączenie. Pełne powiązanie będzie mniej wydajne, ale być może łatwiejsze dadzą dobre wyniki w zależności od danych i zadania. Uważam, że dla 200 000 punktów danych musi to zająć mniej niż sekundę, jeśli używasz odpowiednich struktur danych dla powyższych operacji.
Jak wybrać cechy sieci neuronowej?
Wiem, że nie ma jednoznacznej odpowiedzi na to pytanie, ale załóżmy, że mam ogromną sieć neuronową z dużą ilością danych i chcę dodać nową funkcję na wejściu. „Najlepszym” sposobem byłoby przetestowanie sieci z nową funkcją i zobaczenie wyników, ale czy istnieje metoda sprawdzenia, czy funkcja JEST NIEPRAWDOPODOBNIE pomocna? Podobnie jak miary korelacji (http://www3.nd.edu/~mclark19/learn/CorrelationComparison.pdf) itp.?
Bardzo silna korelacja między nową funkcją a istniejącą funkcją jest dość dobrym znakiem, że nowa funkcja dostarcza niewiele nowych informacji. Prawdopodobnie preferowana jest niska korelacja między nową funkcją a istniejącymi funkcjami. Silna korelacja liniowa między nową cechą a przewidywaną zmienną to dobry znak, że nowa funkcja będzie wartościowa, ale brak wysokiej korelacji nie jest konieczny oznaką słabej cechy, ponieważ sieci neuronowe nie są ograniczone do kombinacji liniowych zmiennych. Jeśli nowy element został utworzony ręcznie z kombinacji istniejących elementów, rozważ pominięcie go. Piękno sieci neuronowych polega na tym, że wymagana jest niewielka inżynieria funkcji i wstępne przetwarzanie – zamiast tego funkcje są uczone przez warstwy pośrednie. O ile to możliwe, preferuj funkcje uczenia się od ich projektowania.
Jak zwiększyć dokładność klasyfikatorów?
Używam przykładu OpenCV letter_recog.cpp do eksperymentowania na losowych drzewach i innych klasyfikatorach. Ten przykład zawiera implementacje sześciu klasyfikatorów – drzewa losowe, wzmocnienie, MLP, kNN, naiwny Bayes i SVM. Używany jest zestaw danych rozpoznawania liter UCI z 20000 instancjami i 16 funkcjami, które dzielę na pół do celów szkoleniowych i testowych. Mam doświadczenie z SVM więc szybko ustawiłem jego błąd rozpoznawania na 3,3%. Po kilku eksperymentach otrzymałem:
Rozpoznawanie liter UCI:
*RTrees – 5.3%
*Boost – 13%
*MLP – 7.9%
*kNN(k=3) – 6.5%
*Bayes – 11.5%
*SVM – 3.3%
Zastosowane parametry:
*RTrees – max_num_of_trees_in_the_forrest=200, max_depth=20,
min_sample_count=1
*Boost – boost_type=REAL, weak_count=200, weight_trim_rate=0.95, max_depth=7
*MLP – method=BACKPROP, param=0.001, max_iter=300 (default values – too slow to experiment)
*kNN(k=3) – k=3
*Bayes – none
*SVM – RBF kernel, C=10, gamma=0.01
Następnie użyłem tych samych parametrów i przetestowałem na zestawach danych Digits i MNIST, najpierw wyodrębniając cechy gradientu (rozmiar wektora 200 elementów):
Cyfry:
*RTrees – 5.1%
*Boost – 23.4%
*MLP – 4.3%
*kNN(k=3) – 7.3%
*Bayes – 17.7%
*SVM – 4.2%
MNIST:
*RTrees – 1.4%
*Boost – out of memory
*MLP – 1.0%
*kNN(k=3) – 1.2%
*Bayes – 34.33%
*SVM – 0.6%
Jestem nowy dla wszystkich klasyfikatorów z wyjątkiem SVM i kNN, dla tych dwóch mogę powiedzieć, że wyniki wydają się dobre. A co z innymi? Spodziewałem się więcej po losowych drzewach, na MNIST kNN daje lepszą dokładność, jakieś pomysły, jak ją zwiększyć? Boost i Bayes dają bardzo niską celność. Na koniec chciałbym użyć tych klasyfikatorów do stworzenia systemu z wieloma klasyfikatorami. Jakakolwiek rada?
Redukcja wymiarowości
Inną ważną procedurą jest porównanie współczynników błędów w szkoleniu i zestawie danych testowych, aby sprawdzić, czy jesteś nadmiernie dopasowany (z powodu „przekleństwa wymiarowości”). Na przykład, jeśli poziom błędów w zestawie danych testowych jest znacznie większy niż błąd w zestawie danych uczących, byłby to jeden wskaźnik. W takim przypadku możesz wypróbować techniki redukcji wymiarowości, takie jak PCA lub LDA. Jeśli jesteś zainteresowany, o PCA, LDA i kilku innych technikach pisałem tutaj: http://sebastianraschka.com/index.html#machine_learning oraz w moim repozytorium GitHub tutaj:
https://github.com/rasbt/pattern_classification
Walidacja krzyżowa
Możesz również przyjrzeć się technikom walidacji krzyżowej, aby ocenić wydajność swoich klasyfikatorów w bardziej obiektywny sposób
Spodziewałem się więcej po losowych drzewach:
* W przypadku lasów losowych, zwykle dla funkcji N, funkcje sqrt (N) są używane do każdej konstrukcji drzewa decyzyjnego. Ponieważ w twoim przypadku N = 20, możesz spróbować ustawić max_depth (liczbę funkcji podrzędnych do skonstruowania każdego drzewa decyzyjnego) na 5.
* Zamiast drzew decyzyjnych zaproponowano i oceniono modele liniowe jako estymatory bazowe w lasach losowych, w szczególności wielomianowej regresji logistycznej i naiwnym modelu Bayesa. Może to poprawić twoją dokładność.
Na MNIST kNN daje lepszą dokładność, jakieś pomysły, jak ją zwiększyć? * Spróbuj z wyższą wartością K (powiedzmy 5 lub 7). Wyższa wartość K dostarczyłaby bardziej wspierających dowodów na temat etykiety klasy punktu.
* Możesz uruchomić PCA lub Linear Discriminant Analysis Fishera przed uruchomieniem najbliższego sąsiada. W ten sposób możesz potencjalnie pozbyć się skorelowanych cech podczas obliczania odległości między punktami, a zatem twoi sąsiedzi k byliby bardziej odporni. Wypróbuj różne wartości K dla różnych punktów w oparciu o wariancję odległości między sąsiadami K.
Grupowanie współrzędnych lokalizacji geograficznej (szer., Długie pary)
Jakie jest właściwe podejście i algorytm klastrowania do klastrowania geolokalizacji? Używam następującego kodu do grupowania współrzędnych geolokalizacji:
importuj numpy jako np
import matplotlib.pyplot as plt
z scipy.cluster.vq import kmeans2, wybielić
współrzędne = np.array ([
[szer. długa],
[szer. długa],
…
[lat, long]
])
x, y = kmeans2 (wybiel (współrzędne), 3, iter = 20)
plt.scatter (współrzędne [:, 0], współrzędne [:, 1], c = y);
plt.show ()
Czy słuszne jest używanie Kmeans do grupowania lokalizacji, skoro wykorzystuje odległość euklidesową, a nie formułę Haversine’a jako funkcję odległości?
W tym przypadku środki K powinny mieć rację. Ponieważ k-średnie próbuje grupować się wyłącznie na podstawie odległości euklidesowej między obiektami, otrzymasz z powrotem skupiska lokalizacji, które są blisko siebie. Aby znaleźć optymalną liczbę skupień, możesz spróbować wykonać „kolanowy” wykres wnętrza
suma grupowa odległości kwadratowej. Może to być pomocne (http://nbviewer.ipython.org/github/nborwankar/LearnDataScience/blob/master/notebooks/D3.%20KMeans%
20Clustering% 20Analysis.ipynb)
K-średnie nie są tutaj najwłaściwszym algorytmem. Powodem jest to, że k-średnie są zaprojektowane tak, aby minimalizować wariancję. To jest oczywiście pojawiające się z punktu widzenia statystyki i przetwarzania sygnałów, ale dane nie są „liniowe”. Ponieważ twoje dane są w formacie szerokości i długości geograficznej, powinieneś użyć algorytmu, który może obsługiwać dowolne funkcje odległości, w szczególności funkcje odległości geodezyjnej. Popularnymi przykładami są klastry hierarchiczne, PAM, CLARA i DBSCAN. Problemy k-średnich są łatwe do zauważenia, gdy weźmie się pod uwagę punkty bliskie zawinięcia + -180 stopni. Nawet jeśli zhakowałeś k-oznacza, aby użyć odległości Haversine, w kroku aktualizacji, gdy ponownie obliczy średnią, wynik będzie źle wkręcony. W najgorszym przypadku k-średnie nigdy się nie zbiegną!
t-SNE Implementacja Pythona: dywergencja Kullbacka-Leiblera
t-SNE, działa poprzez stopniowe zmniejszanie dywergencji Kullbacka-Leiblera (KL), aż do spełnienia określonego warunku. Twórcy t-SNE sugerują użycie dywergencji KL jako kryterium wydajności dla wizualizacji: można porównać rozbieżności Kullbacka-Leiblera, które zgłasza t-SNE. Całkiem dobrze jest dziesięciokrotnie uruchomić t-SNE i wybrać rozwiązanie o najniższej rozbieżności KL [2]. Wypróbowałem dwie implementacje t-SNE:
* python: sklearn.manifold.TSNE ().
* R: tsne, z biblioteki (tsne).
Obie te implementacje, gdy jest ustawiona gadatliwość, wyświetlają błąd (dywergencja Kullbacka-Leiblera) dla każdej iteracji. Jednak nie pozwalają użytkownikowi uzyskać tych informacji, co wydaje mi się nieco dziwne.
Na przykład kod:
importuj numpy jako np
from sklearn.manifold import TSNE
X = np.array ([[0, 0, 0], [0, 1, 1], [1, 0, 1], [1, 1, 1]])
model = TSNE (n_components = 2, verbose = 2, n_iter = 200)
t = model.fit_transform (X)
produkuje:
[t-SNE] Obliczanie odległości parami…
[t-SNE] Obliczone prawdopodobieństwa warunkowe dla próby 4/4
[t-SNE] Średnia sigma: 1125899906842624,000000
[t-SNE] Iteracja 10: błąd = 6,7213750, norma gradientu = 0,0012028
[t-SNE] Iteracja 20: błąd = 6,7192064, norma gradientu = 0,0012062
[t-SNE] Iteracja 30: błąd = 6,7178683, norma gradientu = 0,0012114…
[t-SNE] Błąd po 200 iteracjach: 0,270186
O ile rozumiem, 0,270186 powinno być dywergencją KL. Jednak nie mogę uzyskać tych informacji, ani z modelu, ani z t (co jest prostym numpy.ndarray). Aby rozwiązać ten problem, mógłbym: i) obliczyć dywergencję KL samodzielnie, ii) zrobić coś paskudnego w Pythonie, aby przechwycić i przeanalizować wyjście funkcji TSNE (). Jednakże, ja)
byłoby całkiem głupie ponowne obliczenie dywergencji KL, skoro TSNE () już ją obliczyło, ii) byłoby nieco nietypowe pod względem kodu.
Czy masz jakieś inne sugestie? Czy istnieje standardowy sposób uzyskania tych informacji za pomocą tej biblioteki? Wspomniałem, że wypróbowałem bibliotekę tsne w języku R, ale wolałbym, aby odpowiedzi koncentrowały się na implementacji Python sklearn.
Źródło TSNE w scikit-learn jest w czystym Pythonie. Metoda Fit fit_transform () w rzeczywistości wywołuje prywatną funkcję _fit (), która następnie wywołuje prywatną funkcję _tsne (). Ta funkcja _tsne () ma lokalny błąd zmiennej, który jest drukowany na końcu dopasowania. Wygląda na to, że możesz łatwo zmienić jedną lub dwie linie kodu źródłowego, aby to mieć wartość zwróconą do fit_transform ().
Dlaczego podczas prognozowania powinienem przejmować się danymi sezonowymi?
Mam czas z godzinowym zużyciem gazu. Chcę użyć ARiMR / ARIMA do prognozowania zużycia na następną godzinę na podstawie poprzedniej. Dlaczego powinienem analizować / znaleźć sezonowość (z dekompozycją sezonową i trendową za pomocą Loess (STL)?)?
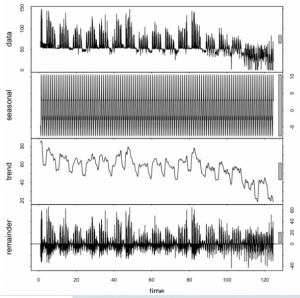
„Ponieważ jest tam”.
Dane mają charakter sezonowy. Więc modelujesz to. Dane mają trend. Więc modelujesz to. Może dane są skorelowane z liczbą plam słonecznych. Więc modelujesz to. Ostatecznie masz nadzieję, że nic nie zostanie do modelowania poza nieskorelowanym szumem losowym. Ale myślę, że spieprzyłeś tutaj obliczenia STL. Twoje reszty wyraźnie nie są nieskorelowane seryjnie. Podejrzewam raczej, że nie powiedziałeś funkcji, że Twoja „sezonowość” to cykl 24-godzinny, a nie roczny. Ale hej, nie podałeś nam żadnego kodu ani danych, więc tak naprawdę nie mamy pojęcia, co zrobiłeś, prawda? Jak myślisz, co w ogóle oznacza tutaj „sezonowość”? Masz jakiś pomysł? Twoje dane wydają się mieć trzy szczyty co 24 godziny. Naprawdę? Czy ten „gaz” = „benzyna” = „benzyna” czy gaz w jakimś układzie ogrzewania / zasilania elektrycznego? Tak czy inaczej, jeśli wiesz a priori, że istnieje 8-godzinny cykl lub 8-godzinny cykl oprócz 24-godzinnego cyklu, który wygląda na bardzo wysoką częstotliwość 1 lub 2-godzinny cykl, umieść to w swoim modelu. Właściwie nawet nie mówisz, jaka jest twoja oś x, więc może jej dni, a wtedy dopasowałbym cykl dzienny, tygodniowy, a potem roczny. Ale biorąc pod uwagę, jak to wszystko zmienia się w czasie = około 85, nie spodziewałbym się, że model poradzi sobie dobrze po obu stronach.
Ze statystykami (a to jest to, przepraszam, że cię zawiodłem, ale nie jesteś jeszcze naukowcem zajmującym się danymi), nie mówisz po prostu robotycznie: „I… Teraz… Dopasuj…… Model S TL…. ”. Patrzysz na swoje dane, próbujesz je zrozumieć, a następnie proponujesz model, dopasowujesz go, testujesz i wykorzystujesz parametry, które wyciąga z danych. Częścią tego jest dopasowanie cyklicznych wzorców sezonowych.
Zalety AUC w porównaniu ze standardową dokładnością
Zacząłem patrzeć na obszar pod krzywą (AUC) i jestem trochę zdezorientowany co do jego użyteczności. Kiedy po raz pierwszy mi wyjaśniono, AUC wydawało się świetną miarą wydajności, ale w moich badaniach odkryłem, że niektórzy twierdzą, że jego przewaga jest głównie marginalna, ponieważ najlepiej nadaje się do łapania “ szczęśliwych ” modeli z wysokimi standardowymi pomiarami dokładności i niskim AUC . Czy powinienem zatem unikać polegania na wartości AUC przy walidacji modeli, czy też kombinacja byłaby najlepsza? Dzięki za całą twoją pomoc.
AUC i dokładność to całkiem różne rzeczy. AUC dotyczy klasyfikatorów binarnych, które wewnętrznie mają pewne pojęcie o progu decyzyjnym. Na przykład regresja logistyczna zwraca wartość dodatnią / ujemną w zależności od tego, czy funkcja logistyczna jest większa / mniejsza niż próg, zwykle domyślnie 0,5. Kiedy wybierasz swój próg, masz klasyfikator. Musisz wybrać jedną. Dla danego progu można obliczyć dokładność, czyli odsetek prawdziwych pozytywów i negatywów w całym zbiorze danych. AUC mierzy, w jaki sposób prawdziwa dodatnia stopa (przypominanie) i fałszywie dodatnia stawka ulegają zmianie, więc w tym sensie mierzy już coś innego. Co ważniejsze, AUC nie jest funkcją progu. Jest to ocena klasyfikatora, ponieważ próg zmienia się we wszystkich możliwych wartościach. W pewnym sensie jest to szerszy miernik, testujący jakość wewnętrznej wartości, którą klasyfikator generuje, a następnie porównuje z wartością progową. Nie jest to testowanie jakości konkretnego wyboru progu. AUC ma inną interpretację, a to oznacza, że jest to również prawdopodobieństwo, że losowo wybrany pozytywny przykład znajdzie się wyżej niż losowo wybrany negatywny przykład, zgodnie z wewnętrzną wartością klasyfikatora dla przykładów.
AUC można obliczyć, nawet jeśli masz algorytm, który tworzy ranking tylko na przykładach. AUC nie jest obliczalne, jeśli naprawdę masz tylko klasyfikator czarnej skrzynki, a nie taki z wewnętrznym progiem. Zwykle dyktują one, który z nich jest w ogóle dostępny dla danego problemu. Myślę, że AUC jest bardziej wszechstronną miarą, chociaż ma zastosowanie w mniejszej liczbie sytuacji. To nie jest lepsze niż dokładność; to jest inne. Zależy to po części od tego, czy bardziej zależy Ci na prawdziwych wynikach, fałszywie negatywnych wynikach, itp. F-miara bardziej przypomina dokładność w tym sensie, że jest funkcją klasyfikatora i jego ustawienia progowego. Ale mierzy precyzję vs zapamiętywanie (prawdziwie dodatni współczynnik), który nie jest taki sam jak w przypadku obu powyższych.
Statystyka + informatyka = nauka o danych?
Chcę zostać naukowcem danych. Studiowałem statystykę stosowaną (nauki aktuarialne), więc mam świetne podłoże statystyczne (regresja, proces stochastyczny, szeregi czasowe, żeby wymienić tylko kilka). Ale teraz mam zamiar zrobić tytuł magistra w dziedzinie informatyki z inteligentnych systemów. Oto mój plan nauki:
*Nauczanie maszynowe
* Zaawansowane uczenie maszynowe
* Eksploracja danych
*Logika rozmyta
* Systemy rekomendacji
* Rozproszone systemy danych
*Chmura obliczeniowa
* Odkrycie wiedzy
* Business Intelligence
*Wyszukiwanie informacji
* Wydobywanie tekstu
Na koniec, przy całej mojej wiedzy statystycznej i informatycznej, czy mogę nazywać siebie naukowcem danych? czy się mylę?
To zależy od tego, do jakiego rodzaju „nauki o danych” chcesz się dostać. W przypadku podstawowych analiz i raportowania statystyki z pewnością pomogą, ale w przypadku uczenia maszynowego i sztucznej inteligencji będziesz potrzebować kilku dodatkowych umiejętności
* Teoria prawdopodobieństwa – musisz mieć solidne podstawy w zakresie czystego prawdopodobieństwa, aby móc rozłożyć każdy problem, widziany wcześniej lub nie, na zasady probabilistyczne. Statystyka bardzo pomaga w przypadku już rozwiązanych problemów, ale nowe i nierozwiązane problemy wymagają głębokiego zrozumienia prawdopodobieństwa, aby można było zaprojektować odpowiednie techniki.
Teoria informacji – to (w stosunku do statystyki) dziedzina całkiem nowa (choć wciąż licząca dziesiątki lat), najważniejsza praca była autorstwa Shannona, ale jeszcze ważniejsza i często zaniedbywana uwaga w literaturze to praca Hobsona, która dowiodła, że Kullback-Leibler Divergence jest jedyną matematyczną definicją, która naprawdę oddaje pojęcie „miary informacji”. Obecnie podstawą sztucznej inteligencji jest możliwość ilościowego określenia informacji.
Teoria złożoności – dużym problemem, z którym boryka się wielu naukowców zajmujących się danymi, który nie ma solidnego tła w teorii złożoności, jest to, że ich algorytmy nie skalują się lub po prostu zajmują bardzo dużo czasu na dużych danych. Weźmy na przykład PCA, ulubioną odpowiedź wielu ludzi na pytanie podczas rozmowy kwalifikacyjnej „jak zmniejszyć liczbę funkcji w naszym zbiorze danych”, ale nawet jeśli powiesz kandydatowi, że „zbiór danych jest naprawdę bardzo duży”, nadal proponują różne formy PCA, które są O (n ^ 3). Jeśli chcesz się wyróżniać, chcesz być w stanie samodzielnie rozwiązać każdy problem, a NIE rzucać na to rozwiązania z podręcznika, które zostało zaprojektowane na długo, zanim Big Data stało się tak modne. W tym celu musisz zrozumieć, jak długo to trwa, nie tylko teoretycznie, ale praktycznie – jak wykorzystać klaster komputerów do dystrybucji algorytmu lub które struktury danych zajmują mniej pamięci. Umiejętności komunikacyjne – ogromną częścią nauki o danych jest zrozumienie biznesu. Niezależnie od tego, czy chodzi o wynalezienie produktu opartego na nauce o danych, czy o dostarczanie informacji biznesowych opartych na nauce o danych, bardzo ważna jest umiejętność dobrej komunikacji zarówno z kierownikami projektów, jak i produktów, zespołami technicznymi i innymi naukowcami zajmującymi się danymi. Możesz mieć niesamowity pomysł, powiedzmy niesamowite rozwiązanie AI, ale jeśli nie możesz skutecznie (a) komunikować DLACZEGO, co przyniesie firmie zyski, (b) przekonać kolegów, że to zadziała oraz (c) wyjaśnić pracownikom technicznym, jak potrzebujesz ich pomocy w jego budowie, a wtedy to się nie uda.
Naukowiec danych (dla mnie) duży termin parasolowy. Naukowca danych postrzegałbym jako osobę, która potrafi biegle posługiwać się technikami z zakresu eksploracji danych, uczenia maszynowego, klasyfikacji wzorców i statystyki. Jednak terminy te są ze sobą powiązane: uczenie maszynowe jest powiązane z klasyfikacją wzorców, a eksploracja danych nakłada się, jeśli chodzi o znajdowanie wzorców w danych. Wszystkie techniki mają swoje podstawowe zasady statystyczne. Zawsze wyobrażam sobie to jako diagram Venna z dużym przecięciem. Informatyka jest również związana z tymi wszystkimi dziedzinami. Powiedziałbym, że do prowadzenia badań komputerowo-naukowych potrzebne są techniki „nauki o danych”, ale wiedza informatyczna niekoniecznie jest implikowana w „nauce o danych”. Jednak umiejętności programistyczne – postrzegam programowanie i informatykę jako różne zawody, w których programowanie jest bardziej narzędziem służącym do rozwiązywania problemów – są również ważne przy pracy z danymi i przeprowadzaniu analizy danych. Masz naprawdę fajny plan nauki i wszystko ma sens. Ale nie jestem pewien, czy „chcesz” nazywać siebie po prostu „naukowcem danych”, mam wrażenie, że „naukowiec danych” to termin tak niejednoznaczny, że może oznaczać wszystko albo nic. Chcę przekazać, że staniesz się kimś więcej – bardziej „wyspecjalizowanym” – niż „tylko” naukowcem zajmującym się danymi.
Czy powinienem wybrać „zrównoważony” zestaw danych, czy „reprezentatywny” zestaw danych?
Moje zadanie „uczenia maszynowego” polega na oddzieleniu nieszkodliwego ruchu internetowego od szkodliwego ruchu. W prawdziwym świecie większość (powiedzmy 90% lub więcej) ruchu internetowego jest niegroźna. Dlatego poczułem, że powinienem korzystać z podobnego rodzaju danych do trenowania moich modeli. Ale potem natknąłem się na jeden lub dwa artykuły badawcze (w mojej dziedzinie), które wykorzystywały wyważone dane do trenowania modeli, sugerując równą liczbę przypadków nieszkodliwego i złośliwego ruchu. Ogólnie rzecz biorąc, jeśli buduję modele ML, czy powinienem wybrać zestaw danych, który jest reprezentatywny dla rzeczywistego problemu, czy też zbalansowany zestaw danych lepiej nadaje się do budowania modeli (ponieważ niektóre klasyfikatory nie zachowują się dobrze w przypadku nierównowagi klas lub z powodu z innych nieznanych mi przyczyn)? Czy ktoś może rzucić więcej światła na zalety i wady obu wyborów oraz jak zdecydować, na który z nich się zdecydować?
Powiedziałbym, że odpowiedź zależy od twojego przypadku użycia. Na podstawie mojego doświadczenia:
* Jeśli próbujesz zbudować reprezentatywny model – taki, który opisuje dane, a nie musi przewidywać – sugerowałbym użycie reprezentatywnej próbki danych.
* Jeśli chcesz zbudować model predykcyjny, szczególnie taki, który działa dobrze według miary AUC lub kolejności rang i planujesz użyć podstawowego frameworka ML (tj. Drzewo decyzyjne, SVM, Naive Bayes itp.), To sugeruję ramy zbalansowanego zbioru danych. W znacznej części literatury na temat nierównowagi klas stwierdza się, że losowe niedopróbowanie (zmniejszanie próby z klasy większości do rozmiaru klasy mniejszościowej) może prowadzić do wzrostu wydajności.
* Jeśli budujesz model predykcyjny, ale używasz bardziej zaawansowanej struktury (tj. Czegoś, co określa parametry próbkowania poprzez opakowanie lub modyfikację struktury pakowania, która próbkuje do równoważności klas), sugerowałbym ponowne podanie reprezentatywnej próbki i pozwalając algorytmowi zająć się zbilansowaniem danych do treningu.
Myślę, że to zawsze zależy od scenariusza. Korzystanie z reprezentatywnego zestawu danych nie zawsze jest rozwiązaniem. Załóżmy, że zestaw treningowy zawiera 1000 negatywnych przykładów i 20 pozytywnych przykładów. Bez jakiejkolwiek modyfikacji klasyfikatora algorytm będzie miał tendencję do klasyfikowania wszystkich nowych przykładów jako negatywnych. W niektórych scenariuszach jest to w porządku. Jednak w wielu przypadkach koszty braku pozytywnych przykładów są wysokie, więc musisz znaleźć na to rozwiązanie. W takich przypadkach można użyć wrażliwego na koszty algorytmu uczenia maszynowego. Na przykład w przypadku analizy danych z diagnostyki medycznej. Podsumowując: błędy klasyfikacyjne nie mają takiego samego kosztu!
Zawsze istnieje rozwiązanie umożliwiające wypróbowanie obu podejść i zachowanie tego, które maksymalizuje oczekiwane wyniki. W twoim przypadku założyłbym, że wolisz zminimalizować fałszywie ujemne wyniki kosztem fałszywie pozytywnych wyników, więc chcesz odnieść swój klasyfikator do silnego negatywu wcześniejszego i zająć się nierównowagą, zmniejszając liczbę negatywnych przykładów w zestawie uczącym. Następnie oblicz dokładność / rozpoznawalność, czułość / specyficzność lub dowolne kryterium, które Ci odpowiada w pełnym, niezrównoważonym zbiorze danych, aby upewnić się, że nie zignorowałeś znaczącego wzorca występującego w rzeczywistych danych podczas budowania modelu na podstawie zredukowanych danych.